
extrarenal MRT of liver
08-06-2022
Extrarenal malignant rhabdoid tumor of liver
Tytułem wstępu
Jeżeli tu jesteś to na pewno masz lub miałeś kogoś z tym rozpoznaniem. Jesteś już po szeregu badań, biopsji lub nawet operacji i szukasz dodatkowych informacji. Niestety brakuje stron zawierających usystematyzowaną wiedzę ukierunkowaną na praktyczne aspekty leczenia – oczywiście jest szereg opracowań angielskojęzycznych, głownie statystycznych pozbawionych jednak istotnych elementów na jakie należy zwrócić uwagę w trakcie leczenia a zwłaszcza w polskich realiach…
Zachęcam również do wymiany informacji lekarzy onkologów, chirurgów, genetyków, którzy chcieli by poszerzyć swoją lub uzupełnić wiedzę zgromadzoną na tej stronie.
Nie jest to opracowanie stricte naukowe, jakikolwiek schemat czy propozycja leczenia a jedynie poradnik dla rodziny pacjenta. Każdy przypadek jest inny i wymaga indywidualnego (bardzo ważne!) podejścia.
Biologia, potencjalne i znane przyczyny
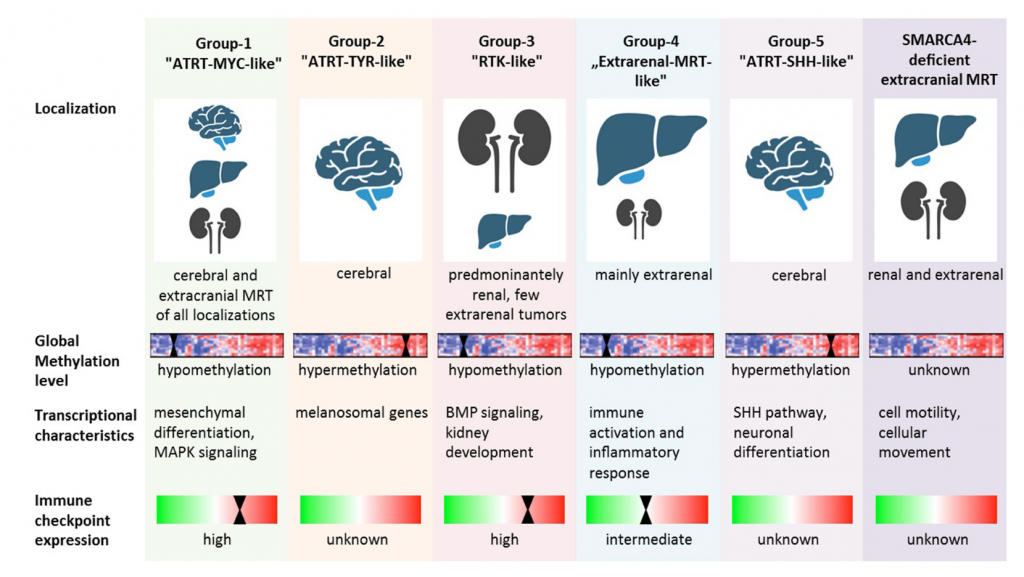
Guzy rabdoidalne (występujące nazwy angielskie RT – rhabdoid tumour, MRT – malignant rhabdoid tumour, extrarenal MRT, RTK – rhabdoid tumor of the kidney, AT/RT – atypical teratoid rhabdoid tumour) to grupa nowotworów występująca głównie u dzieci z największą częstością występowania do pierwszego roku życia – mediana około 11 miesięcy i waha się od 6 miesięcy dla przypadków z dziedziczną (germinalną) zmianą w obrębie SMARCB1 do 1,5 roku dla przypadków bez takiej zmiany – sporadycznych/somatycznych. Późniejsze występowanie jest rzadsze z kilku prostych przyczyn – rokowanie jest dość niekorzystne, przebieg dynamiczny z tendencją do pierwotnego lub późniejszego występowania przerzutów. Generalnie im wcześniejsze pojawienie się niepokojących objawów i wykrycia tym bardziej zjadliwa jest choroba ponieważ jej obraz kliniczny jest szybciej widoczny stąd mniejsza liczba starszych pacjentów. Wedle częstości i lokalizacji występowania wydzielono trzy głowne grupy (Current and Emerging Therapeutic Approaches for Extracranial Malignant Rhabdoid Tumors):
- układ nerwowy – AT/RT (Molecular subgrouping of atypical teratoid/rhabdoid tumors—a reinvestigation and current consensus) – 65% przypadków:
- ATRT-MYC-LIKE (głownie atakuje mózg, wątrobę, nerki i inne lokalizacje poza centralnym układem nerwowym) – transkrypcyjna charakterystyka to: hypometylacja, mezynchymalne różnicowanie, zaburzenie ścieżek sygnałowych MAPK, wysoka ekspresja blokad odpowiedzi układu immunologicznego,
- ATRT-TYR-LIKE (głownie atakuje mózg) – transkrypcyjna charakterystyka to: hypermetylacja, nadekspresja genów melanosomalnych,
- ATRT-SHH-LIKE (głownie atakuje mózg) – transkrypcyjna charakterystyka to: hypermetylacja, nadekspresja ścieżek genów SHH, neuronalne różnicowanie,
- poza układem nerwowym – 35% przypadków,
- RTK (głownie atakuje nerki, niekiedy lokalizacje pozanerkowe – wątrobą, itd.) – transkrypcyjna charakterystyka to: hypometylacja, zaburzenia ścieżek sygnałowych BPK, zaburzenia w rozwoju nerek,
- extrarenal MRT (głownie pozanerkowo – wątroba, tkanki miękkie) – transkrypcyjna charakterystyka to: hypometylacja, immunologiczna aktywacja i odpowiedź zapalna, średnia ekspresja blokad odpowiedzi układu immunologicznego,
- SMARCA4-deficient extracranial MRT (głownie poza centralnym układem nerwowym, wątroba nerki, itd.) – chrakteryzuje się dużą ruchliwością komórek.
Nowotwory RT powstają głownie na bazie mutacji (np. delecji całego genu na skutek reogranizacji chromosomów, usunięcia lub duplikacji całego eksonu, insercji lub delecji porowadzącej do przesunięcia i przedwczesnego zakończenia kodonu, nonsensownych mutacji) somatycznych (70%) lub dziedzicznych (30%) w obrębie genów SMARCB1 (22q11.23) oraz SMARCA4 (19p13.2) w określonym przedziale czasu życia płodowego powodujących zaburzenie w różnicowaniu się komórek. Homozygotyczne delecje są częstsze w guzach pozanerkowych, mutacje missensowne są bardzo rzadkie. W wyjątkowych przypadkach analiza sekwencji kodowania nie wskazuje na przyczynę. W niektórych przypadkach substytucja par zasad w 3-cim nie podlegającym translacji regionie wskazuje na domniemane wyjaśnienie, w przeciwnym wypadku warjacje introniczne prowadzące do nieprawidłowego wstawienia pseudooksonu w czasie transkrypcji może doprowadzić do pełnej dezaktywacji SMARCB1.
Jak powyżej podejrzewa się w nietypowych przypadkach zaburzenia transkrypcji, epigenetyczne lub mieszaną budowę:
- niezbadany mechanizm utraty INI1 posttranskrypcyjny nie wynikający z utraty w materiale genetycznym – https://pubmed.ncbi.nlm.nih.gov/22020042/
- budowa mieszana guzów zawierająca w różnych proporcjach komórki zachowujące lub nie INI1 – https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2743242/
- dodatkowy podtyp guzów RT zachowujący INI1 ale mający wszelkie cechy i zbliżoną przeżywalność do RT – https://bmcmedgenomics.biomedcentral.com/articles/10.1186/s12920-015-0103-3
Właściwie wszystkie całkowite delecje genu SMARCB1 lub większe germinalne delecje 22q11.2 zawierające SMARCB1 są de novo (powstają w komórce rozrodczej lub są przekazane przez rodziców), to samo tyczy się mutacji germinalnych SMARCB1. Około 20% germinalnych zmian są delecjami w obszarze 22q11.2 chromosomu, natomiast 25% pacjentów posiada częściową delecję lub duplikację obejmującą 1-5 exonów. Pozostali pacjenci mają różnorodne mutacje skracające spowodowane punktowymi mutacjami lub podstawieniami/delecjami prowadzącymi do przesunięcia ramki odczytu. Mutacje miejsca splicingowego są najmniej powszechne.
Najczęstsze mutacje germinalne SMARCB1 dotyczą eksonów 2 i 4. Z wyjątkiem mutacji przesunięcia ramki na eksonie 9-tym te same mutacje predysponują do AT/RT, RTK i w mniejszym stopniu do eMRT. Większość przypadków eMRT jest sporadyczna i powstaje jako homozygotyczna utrata SMARCB1 spowodowana delecją, niezbalansowanymi translokacjami 22q11.2 lub monosomią 22. Najczęstszą przyczyną drugiego uderzenia (teoria Knudsona) u pacjentów z germinalnymi mutacjami jest duża delecja obszaru 22q lub monosomia 22, neutralna pod względem kopii utrata heterozygotyczności (CN-LOH) powodująca uwidocznienie mutacji lub delecji na pozostałej alleli. Duża liczba (43%) guzów posiada mutację na jednej alleli a druga kopia genu ulega utracie na skutek powyższych przyczyn. Złożone heterozygotyczne mutacje są rzadkie u tych pacjentów (4%). Częściowe delecje i duplikacje są wykrywane w 15% przypadków. Homozygotyczne delecje eksonów 1-9 SMARCB1 ujawniają się w 40% ogólnej liczby RT z niejednorodną dystrybucją pomiędzy lokalizacjami anatomicznymi (25% AT/RT, 40% RTK, 70% eMRT).
Mutacje w sporadycznych RT obejmują punktowe mutacje pojedyńczych par zasad, insercje/delecje lub mutacje przesunięcia ramki odczytu, które są posądzane o wprowadzanie kodonu stopu. Duża liczba mutacji sprowadza się do NMD (ang. Nonsense-Mediated Decay) jednakże nie jest to formalnie udowodnione w większości przypadków. Najszęstsza częstotliwość mutacji sekwencji kodonów występuje na eksonie 9-tym. Delecje kodonów 381 oraz 382 są wyłącznie związane z AT/RT. Mutacje na eksonach 2 i 4-7 są często obserwowane w RTK i AT/RT. Niektóre specyficzne mutacje na eksonach 2, 4, i 5 powtarzają się bardzo często ale nie wydają się być specyficzne na lokalizacji w mózgu czy nerkach. Mutacje na eksonach 1 i 3 są bardzo rzadkie a na eksonie 8 zostały udokumentowane na jednym przypadku RT. Mutacje splicingowe są wyjątkowe w pierwotnym RT a mutacje missenowne nie zostały dotychczas zaraportowane.
Utrata SMARCB1 odpowiada również za powstawanie schwanomatozy – rodzaju neurofibromatozy, która może się przekształcić w złośliwe mięsaki.
Przekrojowy przegląd literatury:
- Somatic mutations and single-cell transcriptomes reveal the root of malignant rhabdoid tumours
- Biology and Treatment of Rhabdoid Tumor
- Extracranial rhabdoid tumours: what we have learned so far and future directions
- In vitro Modeling of Embryonal Tumors

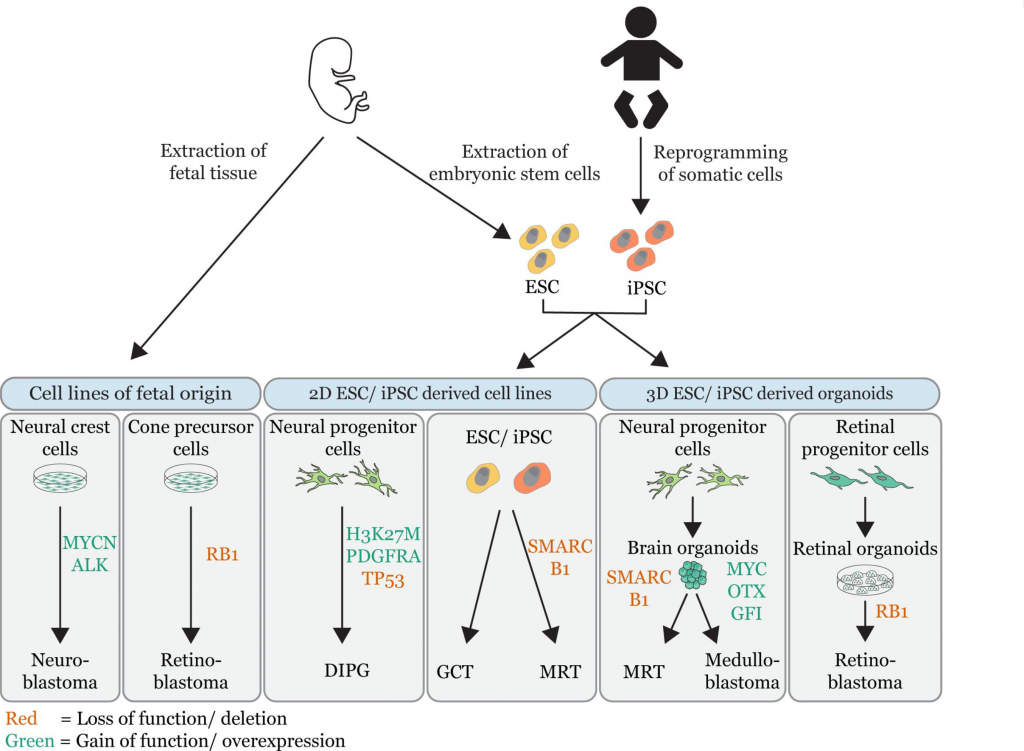
Aktualnie zakłada się, że źródłem RT jest linia prymitywnych protoplastycznych/prekursorowych komórek macierzystych (pluripotent stem cells PCS takich jak embryonic stem cells ESCs i induced PSCs iPSCs – reprogramowane komórki somatyczne) ze zdolnością do rożnorodnego różnicowania i prawdopodobnie wywodzi się z komórek grzebienia nerwowego (neural crest). Komórki te mogą się samoodnawiać i mogą byc poddane różnicowaniu do wszystkich rodzaji lisków/warstw komórek zarodkowych (germ layers tj. endoderm, ectoderm, mesoderm, neural crest).
Znacząca grupa nowotworów wieku dziecięcego (pediatrycznych) objawia się we wczesnym dzieciństwie sugeruje to ich początek w rozwoju prenatalnym. Te tak zwane nowotwory embrionalne uważane są jako konsekwencja anormalnego rozwoju. Jendakże wiele z tych nowotworów ma nieustaloną przyczynę. Nowotwory osób dorosłych rozwijają się poprzez sukcesywną akumulację mutacji przez wiele lat, natomiast nowotwory embrionalne typowo charakteryzują się nieznaczym obciążeniem mutacjami i niewielką ilością zdarzeń genetycznych aby zainicjować nowotworzenie. Ta niewielka zmienność genetyczna, która w nich zachodzi powoduje jendakże, że te komórki płodowe zachowują podobny do prekursorowego stan i nie podlegają różnicowaniu. Ta blokada dojrzewnia popycha te komórki do złośliwej transformacji. Odkrycia te wskazują, że samo zachowanie wczesnego embrionalnego kotekstu komórkowego jest wystarczające do zapoczątkowania nowotworu.
Dominującą grupą genów podlegających mutacji w dziecięcych jak i dorosłych nowotworach są podjednostki SWItch/Sucrose Non-Fermentable (SWI/SNF) kompleksu remodulującego chromatynę (pozostałe kompleksy to CHD i INO).
Kompleks ten składa się z genów odpowiadających za konkretne fenotypy i nowotwory Pharmacoepigenetics:
- SMARCA1-(4)-5: SFMS, schizofrenia, opóżnienie intelektualne związane z chromosomem X, rak żołądka, NBS, rak płuc, rak jelita grubego, rak macicy, CSS4, kardiomiopatia przerostowa, rak prostaty, RTPS2, RT, rak piersi,
- SMARCAD1: adermatoglifia, BS, rak piersi,
- SMARCAL1: SIOD,
- SMARCB1: CML, ES, rodzinna Schwannomatoza, oponiak, CSS3, RT somatyczny, RTPS1,
- SMARCC1-2: rak prostaty, rak jelita grubego, rak żołądka,
- SMARCD1-3: rak żołądka, rak jajników, SGD2, zapalenia,
- SMARCE1: CSS5, oponiak, rak prostaty.
Rola oddziaływania tego kompleksu jest bardzo wyraźna w MRT charakteryzując się utratą SMARCB1/SMARCA4. Badania na modelach komórkowych jasno dowodzą, że utrata SMARCB1 indukuje powstanie nowotworu rabdoidalnego w bardzo ograniczonym przedziale czasu i dla wybranych linii/typów komórek płodowych. Zmodyfikowane (usunięty gen SMARCB1) komórki iPCS rozwineły MRT, te komórki które dodatkowo były w stanie rozwinąć się w neural progenitor cells (NPCs) wytowrzyły inne nowotwory ale bez cech rabdoidalnych co oczywiście tłumaczy zróżnicową budowe RT.
Oddzielny poziom epigenetycznych regulacji pojawiający się w nowotworach embrionalnych to postranslacyjne modyfikacje zakończeń histonów, które nadzorują przejście pomiędzy wyzwalającymi i hamującymi markerami histonowymi odpowiadającymi za dynamiczną regulację ekspresji genów w czasie rozwoju osobniczego – szeroko opisane występowanie w diffuse intrinsic pontine glioma (DIPG).
Podział komórek jest złożonym procesem biologicznym, w którym wiele etapów może odbyć się nieprawidłowo – na przykład błędy polimerazy DNA w czasie replikacji, stochastyczne nieprawidłowości w dystrybucji komponentów wśród komórek potomnych. Komórki podlegające podziałom są narażone na działanie czynników genotoksycznych oraz innych niszczących endo i ekso gennych. Jest dowiedzione, że pewne tkanki mają tendencje do powstawania nowotworów milion razy częściej niż pozostałe – odpowiada za to ilość podziałów normalnych komórek macierzystych w tych tkankach. Nieprawidłowości w komponentach eliminujących zanieczyszczenia nie tylko powodują uszkodzenia komórkowe ale także mogą aktywować podziały komórkowe, które niekontrolowane w większej ilości prowadzą do błędów replikacji DNA. Komórki macierzyste pozostając w niszach są w nich chronione oraz regulują ich procesy biologiczne takie jak proliferacja i tendencja do migracji. Zaburzenie tych procesów może prowadzić do rozwoju nowotworu. Na przykład urazy tkanek są znanym czynnikiem aktywującym podział komórek w celach naprawczych a obserwacje i badania eksperymentalne wiążą urazy z nowotworami.
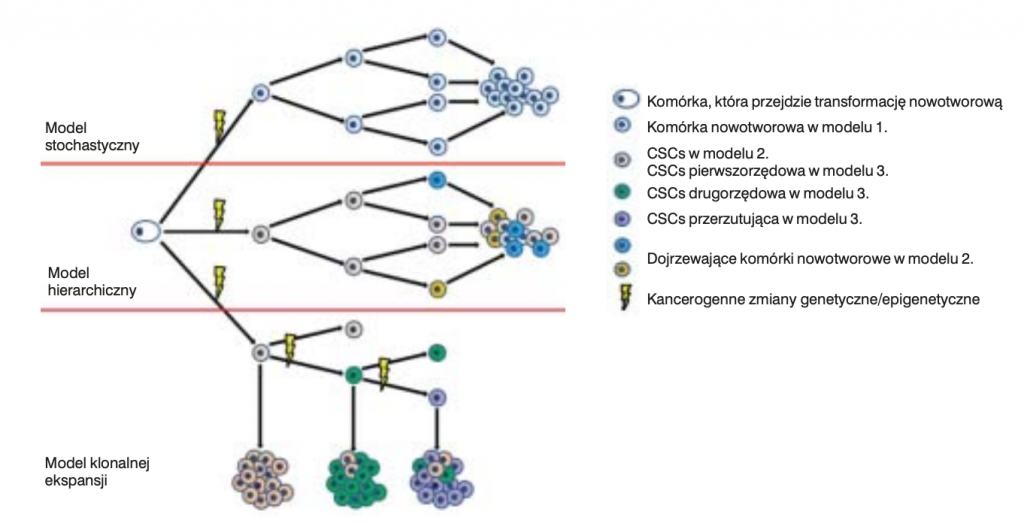
Istnieje teoria nowotworowych komórek macierzystych – subpopulacji komórek nowotworowych mająca nadzwyczajne możliwości samoodnawiania się, rozsiewu, chemo – radio odporności. Teoria ta ogólnie zakłada:
- komórki nowotworowe powstają z normalnych komórek macierzystych,
- głowną przyczyną powstawania nowotworów jest powstanie uszkodzeń w komórkach macierzystych w czasie ich podziałów,
- skumulowanie wielu uszkodzeń w komórkach macierzystych powoduje powstawanie nowotworowych komórek macierzystych (cancer stem cells CSC),
- przerzuty powstają kiedy komórki macierzyste (rozregulowane, przednowotworowe albo nowotworowe) albo ich nowotworowe komórki potomne opuszczją ich naturalną tkankę (niszę) – niekoniecznie guza pierwotnego – i formują nowe guzy w innych lokalizacjach.
Hipoteza nowotworowych komórek macierzystych sugeruje, że niewielka populacja komórek jest zdolna do otworzenia całego nowotworu. Zgodnie z tym modelem powodem, dla którego nowotwór jest zdolny do samoodtworzenia są komórki macierzyste – samoodnawialne i zdolne do różnicowania się w kilka typów komórek. Leki przeciwnowotworowe/radioterapia działają na szybko dzielące się komórki stanowiące większą część masy guza, natomiast przynoszą niewielką szkodę komórkom macierzystym.
Ze względu na potencjał różnicowania komórek macierzystych można wyróżnić:
- komórki totipotencjalne – mogą dać początek wszystkim wyspecjalizowanym komórkom w czasie prawidłowej embriogenezy,
- komórki pluripotencjalne – różnicują się do wielu ale nie wszystkich typów komórek ciała powstających ze wszystkich trzech listków zarodkowych,
- komórki multipotencjalne – różnicują się do komórek specyficznych dla danej niszy – środowiska tkankowego lub narządu,
- komórki unipotencjalne – różnicują się tylko na jedną linię komórkową.
Sądzi się, że komórki macierzyste występują w większości tkanek ciała dorosłego człowieka, jako tak zwane tkankowe (somatyczne) komórki macierzyste. W większości tkanek tworzą one bardzo małą populację, najczęściej stanowiąc tylko 1–2% wszystkich komórek, lecz ich obecność wydaje się niezbędna w czasie całego życia pozapłodowego. Komórki macierzyste osób dorosłych (adult stem cells), poprzez asymetryczne podziały tworzą odpowiednią liczbę komórek zróżnicowanych, które uzupełniają pulę komórek wyspecjalizowanych. Druga z komórek potomnych, powstających wskutek podziału asymetrycznego, pozostaje niezróżnicowaną komórką macierzystą o identycznych właściwościach jak ta, z której powstała. Asymetryczny podział komórek macierzystych służy więc zarówno do odnowienia ich puli, jak i do zapoczątkowania drogi różnicowania do określonego typu komórek. W warunkach prawidłowych komórki macierzyste dzielą się rzadko. Niewielka aktywność proliferacyjna pełni funkcję mechanizmu chroniącego komórki przed wyczerpaniem się ich puli i przed mutacjami, jakie często pojawiają się w czasie replikacji.
Zakłada się, że nowotworowe komórki macierzyste mogą stanowić 1-80% komórek danego guza. Udowodniono też, że ilość CSC w guzie na początku leczenia jest odwrotnie proporcjonalna do wyników leczenia. Tak więc dezaktywacja CSC w guzie jest celem skutecznego leczenia.
Nisza jest to specyficzne mikrośrodowisko, w którym są zlokalizowane komórki danego typu. W jej skład wchodzą komórki, składniki macierzy pozakomórkowej oraz czynniki rozpuszczalne, takie jak cytokiny lub czynniki wzrostu, które mogą być również wydzielane na drodze auto czy parakrynnej przez komórki danej niszy. Warunki, jakie panują w obrębie niszy, regulują podziały, różnicowanie, angiogenezę oraz tworzenie przerzutów przez CSCs. Komórki te odpowiadają na sygnały generowane przez wszystkie elementy danego mikrośrodowiska i dodatkowo są w stanie je modyfikować, tworząc środowisko optymalne dla swego rozwoju. Warunki mikrośrodowiska mają ogromne znaczenie dla utrzymania odpowiedniej puli CSCs w stanie niezróżnicowanym, podobnie jak to jest w przypadku somatycznych komórek macierzystych w niszach tkankowych. Oprócz tego, jak pokazały wyniki badań, zmiany w obrębie niszy mogą indukować proces nowotworzenia lub wręcz przeciwnie – hamować go, nawet w przypadku nagromadzenia się zmian proonkogennych. Nisza może kontrolować procesy nowotworzenia między innymi poprzez kontrolę ekspresji genów (mechanizmy epigenetyczne). W warunkach doświadczalnych zademonstrowano, że jeżeli somatyczne komórki macierzyste wyizoluje się, to poza swoją fizjologiczną niszą tracą one swoje normalne funkcje i nabywają właściwości kancerogennych. Wykazano, że w wyniku istnienia przewlekłego stanu zapalnego zmianie może ulec mikrośrodowisko, co w konsekwencji może prowadzić do rozwoju nowotworu, jak to ma miejsce na przykład w przypadku raka jelita grubego w przebiegu nieswoistych zapaleń jelita grubego. Ogromne znaczenia dla funkcjonowania
CSCs w danej niszy ma stan hipoksji (niedotlenienia), jaki tam panuje i jaki jest aktywnie wywoływany i utrzymywany przez komórki nowotworowe. W warunkach fizjologicznych jedynie komórki macierzyste, spośród wszystkich prawidłowych komórek ciała, potrzebują do swojego prawidłowego funkcjonowania warunków niedotlenienia, co stanowi kolejne podobieństwo CSCs do tkankowych komórek macierzystych. Hipoksja wywołuje wiele procesów, które są indukowane dzięki aktywacji czynnika transkrypcyjnego HIF (hypoxia inducible factor). W badaniach doświadczalnych wykazano, że CSCs dzięki czynnikowi HIF zwiększają progresję choroby. Czynnik ten reguluje ekspresję genów wielu białek kluczowych dla funkcjonowania komórek, dzięki którym sprawnie one reagują i odpowiadają na zmiany stężenia parcjalnego tlenu w otoczeniu. Między innymi HIF odpowiada za aktywację genów związanych
z przełączeniem metabolizmu na beztlenowy, bardziej wydajny transport glukozy do wnętrza komórek oraz indukcję angiogenezy. Większość komórek może w stanie prawidłowym funkcjonować tylko w obrębie swojej niszy, podczas gdy CSCs i ich komórki potomne są w stanie kreować dogodne dla swojego rozwoju warunki mikrośrodowiska i potrafią przetrwać w innych, nietypowych dla siebie miejscach organizmu, w tak zwanych niszach przedprzerzutowych (premetastatic niche). Tam adaptują się do istniejących warunków, by następnie dostosować je do swoich potrzeb, co jest niezbędne dla rozwoju nowego guza w miejscu, dokąd przedostały się w trakcie tworzenie przerzutów. Dodatkowo, istotną rolę w obrębie niszy nowotworowych odgrywa swoiste mikrośrodowisko immunologiczne w obrębie guza, które cechuje immunosupresja, czyli wyciszenie aktywności komórek układu odpornościowego.
Uważa się, że CSCs mogą się rozwijać z prawidłowych komórek macierzystych lub powstających z nich częściowo zróżnicowanych komórek (progenitorowych) obecnych w danej niszy lub też mogą pochodzić od w pełni zróżnicowanych komórek somatycznych. Kwestii tej nie można rozstrzygnąć w sposób jednoznaczny dla większości znanych typów nowotworów. Prawidłowe tkankowe komórki macierzyste mają długi czas życia, więc może w nich dojść do nagromadzenia wystarczającej liczby mutacji, by przejść transformację nowotworową. Posiadają one zdolność do podziałów i potencjał do różnicowania się, więc łatwiej im osiągnąć „nieśmiertelność” (czyli zdolność do niekontrolowanych podziałów) i wymknąć się spod kontroli białek regulujących cykl komórkowy.
Stosowane obecnie środki terapeutyczne zawodzą w wielu przypadkach różnych rodzajów nowotworów, ponieważ nie działają na CSCs. Terapia jest stosunkowo skuteczna wobec proliferujących lub różnicowanych komórek guza, natomiast z reguły jest nieefektywna wobec komórek odpowiedzialnych za rozwój i progresję nowotworu. Zaobserwowano, że stosowanie niektórych terapeutyków powoduje selekcję komórek i znaczny wzrost względnego udziału CSCs wśród komórek guza. Oznacza to,
że CSCs charakteryzują się rzeczywiście większą odpornością na działanie chemioterapeutyków. Dodatkowo, zaobserwowano, że CSCs potrafią przeprowadzać fuzje z komórkami somatycznymi danej niszy. Takie tak zwane chimeryczne komórki są bardziej agresywne i bardziej odporne na terapię.
Problem w opracowaniu strategii walki z CSCs polega na tym, że ich identyfikacja oparta jest głównie na markerach fenotypowych (obecności pewnych białek powierzchniowych), które dla każdego typu nowotworu mogą być różne. Nie jest znany jeden uniwersalny marker CSCs.
W przypadku nowotworów, gdzie pierwszą i podstawową metodą terapii jest chirurgiczne usunięcie zajętego fragmentu, należy pamiętać, że CSCs mogły już się oderwać od guza pierwotnego (dzięki swoim dużym skłonnościom do migracji) i osiąść w innych miejscach. Z dużym prawdopodobieństwem staną się one później źródłem regresji choroby. Z tego powodu wysiłek badaczy obecnie jest skierowany na opracowanie metod terapii uzupełniającej, która byłaby swoista względem mikroprzerzutów niewykrywalnych przy użyciu znanych metod diagnostycznych.
Szerszy opis:
- Rola nowotworowych komórek macierzystych w patogenezie i terapii chorób nowotworowych
- https://biotechnologia.pl/biotechnologia/nowotworowe-komorki-macierzyste,17296
- Stem cell division theory of cancer
- Cancer stem cell theory: Are we moving past the mist?
- In Vivo Expansion of Cancer Stemness Affords Novel Cancer Stem Cell Targets: Malignant Rhabdoid Tumor as an Example
- Human Pluripotent Stem Cell-Derived Tumor Model Uncovers the Embryonic Stem Cell Signature as a Key Driver in Atypical Teratoid/Rhabdoid Tumor
- Neural Crest and Cancer: Divergent Travelers on Similar Paths
Co ciekawe istnieje pewna korelacja lub zwykła zbieżność pomiędzy RT a występowaniem syndromu DiGeorge-a z uwagi na występowanie predysponujących mikrodelecji w tym samym obszarze chromosomu 22 – 22q11.2.
- The recurrent distal 22q11.2 microdeletions are often de novo and do not represent a single clinical entity: a roposed categorization system – https://rhabdoid.info/wp-content/uploads/2021/12/gim201379.pdf
Guzy AT/RT pomimo swego podobieństwa histologicznego, klinicznego, występujących podobnych mutacji genetycznych posiadają dodatkowe właściwości oraz dedykowane sposoby leczenia (inne drogi podawania chemioterapeutyków), że wydziela się je do osobnych opracowań. Aktualnie największe nadzieje w tym rozpoznaniu daje postępowanie oparte o MEMMAT lub MUV-ATRT (Austria) zwłaszcza dla nawrotów lub braku skuteczności bieżącego leczenia.
- Antiangiogenic Therapy for Children With Recurrent Medulloblastoma, Ependymoma and ATRT (MEMMAT) – https://clinicaltrials.gov/ct2/show/NCT01356290
- Atypical teratoid rhabdoid tumor: improved long-term survival with an intensive multimodal therapy and delayed radiotherapy. The Medical University of Vienna Experience 1992–2012 – https://rhabdoid.info/wp-content/uploads/2021/12/Atypical_teratoid_rhabdoid_tum.pdf
Z uwagi na to, że nowotwór ten może zlokalizować się praktycznie w każdym miejscu ciała wymaga ostrożnego różnicowania względem nowotworów występujących w danej lokalizacji i wywodzących się z tkanek tej lokalizacji. Cechą łącząca jest utrata SMARCB1 (INI1, 95% przypadków) lub SMARCA4 (BRG1, 5% przypadków) najczęściej wykrywana w czasie badania histopatologicznego IHC (immunohistochemicznego) próbki guza objawiająca się brakiem INI1 dla pierwszego z nich. Utraty BRG1 nie wykonuje się rutynowo dlatego wykrycie tego wariantu jest możliwe dopiero w trakcie badania molekularnego/genetycznego. W razie występowania mutacji w obrębie wspomnianych genów mogą występować synchroniczne guzy pierwotne w tej samej lub innych lokalizacjach sugerujące występowania zespołu predyspozycji – RTPS (Rhabdoid Tumor Predisposition Syndrome). Takie podejrzenie wymaga dokładnego przebadania wszystkich członków najbliższej rodziny celem oceny możliwości występowania u nich choroby w przyszłości. W obrazie histopatologicznym występują komórki drobne rhabdoidalne, wrzecionowate epitheloidne na myksoidalnym podścielisku. Odczyny: desmin (-), MyoD1(-), vimentin (+). Może pojawiać się także dodatni odczyn GPC3 (Glypican-3), który może potencjalnie wskazywać na możliwość stosowania terapii CAR-T ale wymagana jest jego dokładna ocena ilościowa i gęstości (>25%, >2)
- Immunotherapeutic Targeting of GPC3 in Pediatric Solid Embryonal Tumors – https://www.frontiersin.org/articles/10.3389/fonc.2019.00108/full
- Interleukin-15 Armored Glypican 3-specific Chimeric Antigen Receptor Expressed in T Cells for Pediatric Solid Tumors – https://clinicaltrials.gov/ct2/show/NCT04377932.
Nowotwór zlokalizowany w wątrobie należy do kategorii mięsaków non-RMS (Rhabdomyosarcoma) like natomiast jego leczenie powinno być tak intensywne jak RTK (nerki) lub nawet bardziej intensywne. Wątroba jest organem pojedyńczym o kluczowej funkcji dla organizmu i niekorzystnym centralnym położeniu wśród kluczowych naczyn krwionośnych, zewnętrznych dróg żółciowych, trzustki, żołądka, jelit, wszelkie krytyczne nieprawidłowości w jej działaniu spowodują brak możliwości dlaszego leczenia dlatego należy zmaksymalizować leczenie zanim te niekorzystne warunki wystąpią. Lokalizacja często początkowo sugeruje nowotwór wywodzący się z komórek wątroby (drobnokomórkowa niezróżnicowana hepatoblastoma SCUD – Small Cell Undifferentiated) zwłaszcza gdy towarzyszy temu wstępnemu rozpoznaniu podwyższenie specyficznego markera AFP (alfa-fetoproteina) lub neuroblastomę gdy badania obrazowe nie są w stanie jednocześnie wskazać czy guz wywodzi się czy tylko nacieka wątrobę a jednocześnie jest podwyższony marker NSE (Neuron-Specific Enolase). Bywają też możliwe różnicowania względem proksymalnego typu mięsaka epiteloidnego (proximal-type ES Proximal-Type Epithelioid Sarcoma). Różnicowanie musi być bardzo ostrożne i wykonywane w specjalistycznym ośrodku.
- Nowotwory tkanek miękkich – polskie opracowanie – https://rhabdoid.info/wp-content/uploads/2021/12/Miesakitkanekmiekkichmlodzi.pdf
- Malignant Rhabdoid Tumor, an Aggressive Tumor Often Misclassified as Small Cell Variant of Hepatoblastoma – https://rhabdoid.info/wp-content/uploads/2021/12/cancers-11-01992.pdf
- Expression of CD34 and β-Catenin in Malignant Rhabdoid Tumor of the Liver Mimicking Proximal-Type Epithelioid Sarcoma https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5964282/
- Cuda
- Cuda niestety się nie zdarzają – w trakcie leczenia na pewno dojdzie do różnych nieprzewidzianych zdarzeń oraz ich wzajemnych oddziaływań, które można odczytać jako noszące znamiona jakiejś zewnętrznej interwencji natomiast nic się cudownie nie uleczy.
Cuda trzeba sobie zorganizować:- Być w odpowiednim ośrodku.
- Uzbroić się w odpowiednią wiedzę.
- Zadawać odpowiednie pytania.
- Działać metodycznie i na wielu płaszczyznach.
- Planować z wyprzedzeniem.
- Zakładać zawsze najgorszy wariant.
- Cuda niestety się nie zdarzają – w trakcie leczenia na pewno dojdzie do różnych nieprzewidzianych zdarzeń oraz ich wzajemnych oddziaływań, które można odczytać jako noszące znamiona jakiejś zewnętrznej interwencji natomiast nic się cudownie nie uleczy.
- Ośrodek – Diagnostyka wstępna
- W tym momencie można założyć, że w ramach różnicowania jest już po biopsji lub operacji i histopatologicznym rozpoznaniu RT dlatego tematyka tego zagadnienia będzie rozbudowywana później.
- W tym momencie można założyć, że w ramach różnicowania jest już po biopsji lub operacji i histopatologicznym rozpoznaniu RT dlatego tematyka tego zagadnienia będzie rozbudowywana później.
- Rozpoznanie
- Rozpoznanie dokonywane jest na podstawie analizy histopatologicznej w tym IHC próbek tkanki guza gdzie odnajdowane są rabdoidalne typu komórek oraz swoiste antygeny w tym kluczowy INI1 oraz BRG1. Guz może zostać też poddany badaniu molekularnemu stwierdzającemu mutację (np. delecję) w obrębie genów SMARCB1 / SMARCA4.
- Rozpoznanie dokonywane jest na podstawie analizy histopatologicznej w tym IHC próbek tkanki guza gdzie odnajdowane są rabdoidalne typu komórek oraz swoiste antygeny w tym kluczowy INI1 oraz BRG1. Guz może zostać też poddany badaniu molekularnemu stwierdzającemu mutację (np. delecję) w obrębie genów SMARCB1 / SMARCA4.
- Ośrodek – Leczenie
- Prawdopodobnie biopsja została wykonana w szpitalu gdzie była wykonywana wstępna diagnostyka lub bardziej specjalistycznym ośrodku (Gdańsk – UCK Uniwersyteckie Centrum Kliniczne, Warszawa – CZD Centrum Zdrowia Dziecka). Jesteście w trakcie rekonwalescencji po operacji już z wynikiem histopatologicznym lub w trakcie dalszego leczenia. W tym momencie musicie upewnić się, że jest to ośrodek referencyjny wielospecjalistyczny:
- Posiadający kwalifikacje do leczenia mięsaków tkanek miękkich – tj. mający minimum 25 takich przypadków rocznie.
- Posiadający chirurgię dziecięcą specjalizującą się w chirurgii wątroby.
- Posiadający możliwość przeszczepu wątroby.
- Posiadający lub współpracujący z pobliskim referencyjnym ośrodkiem radioterapii.
- Posiadający oddział onkologiczny zajmujący się czynnie przypadkami RT.
- Posiadający międzynarodowe kontakty i doświadczenie.
Jedynym znanym mi ośrodkiem w Polsce spełnijącym wszystkie te kryteria razem jest Centrum Zdrowia Dziecka w Warszawie.
- Niezbędne badania po rozpoznaniu:
- Badania IHC ekspresji białek w poszukiwaniu odpowiedzi na standardowe chemioterapeutyki stosowane w leczeniu RT i innych nowotworw oraz immunoterepeutyki (o tym dalej w leczeniu „niestandardowym”).
- Genetyczne badanie krwi obwodwej i guza (dla potwierdzenia) na występowanie mutacji SMARCB1 / SMARCA4. W przypadku wykrycia mutacji w krwi obwodowej rokowanie jest jeszcze gorsze, potrzebna jest dalsza diagnostyka w kierunku występowania guzów sychnronicznych. Należy przebadać też pozostałych członków najbliższej rodziny (rodziców, rodzeństwo) w kierunku występowania RTPS (RPTS1 i RPTS2) i dalszej oceny rokowniczej dla tych osób (https://rhabdoid.info/wp-content/uploads/2022/05/Frühwald2021_Article_CurrentRecommendationsForClini.pdf).
- Molekularne poszukiwanie w DNA guza celów molekularnych dla zastosowania terapii celowanej i wykrycia ścieżek sygnałowych sprzyjających powstawaniu nowotworów.
- Badanie poziomów metylacji.
- Prawdopodobnie biopsja została wykonana w szpitalu gdzie była wykonywana wstępna diagnostyka lub bardziej specjalistycznym ośrodku (Gdańsk – UCK Uniwersyteckie Centrum Kliniczne, Warszawa – CZD Centrum Zdrowia Dziecka). Jesteście w trakcie rekonwalescencji po operacji już z wynikiem histopatologicznym lub w trakcie dalszego leczenia. W tym momencie musicie upewnić się, że jest to ośrodek referencyjny wielospecjalistyczny:
- Chemioterapia
- Zanim dojdziemy do konkretnych „protokołow” i ich nazw należy jasno powiedzieć, że jest to tylko schemat postępowania, zbiór dobrych praktyk, konsensus, opracowanie historycznych doświadczeń grup badawczych, złoty środek. W żadnym przypadku nie może być on traktowany dosłownie a zwłaszcza przez ośrodek nie mający do czynienia z takimi przypadkami na co dzień. Każdy przypadek należy traktować indywidualnie, dostosowująć w jak najlepszej wierze do określonego pacjenta, jego aktualnych i przyszłych potrzeb zapewniając wzrost szans na długie przeżycie. W dokumentach tych jest tak wiele niuansów, powtórzeń, znaków zapytania wynikających z prostego faktu – przypadków jest mało, rozpoznanie zwłaszcza historyczne mogło być wstępnie nieprawidłowe ze względu na trudność w rożnicowaniu, dość dynamiczny przebieg u młodszych pacjentów, możliwość występowania praktycznie w całym organiźmie, dobór środków i narzędzi leczniczych – zatem trudno wyróżnić jeden konkretny działający schemat leczenia w każdym przypadku a trudno zastosować taki ogólny plan bez jego precyzyjnego indywidualnego dostosowania.
Jednym z takich rejestrów/”protokołów” jest europejski EU-RHAB (2009 r.). Jest to rejestr przypadków oraz zbiór rekomendacji na zasadzie konsensusu powstały na podstawie analizy danych historycznych, własnych, grup GPOH i SIOP oraz współpracy z EpSSG i COG. Wszelkie decyzje w przypadku posługiwania się tym „protokołem” należą tylko i wyłącznie do lekarza prowadzącego. Stąd ogromna potrzeba prowadzenia leczenia przez wykwalifikowany wielospecalistyczny zespół w referencyjnej wielospecjalistycznej placówce.
Inny moim zdaniem bardziej ukierunkowany na klasyfikację rozpoznania i samą lokalizację to protokół EpSSG NRSTS zakładający maksymalizację dawkowania wraz z minimalizacją odstepu między cyklami, zkładający wczesną radioterapię i wskazujące konkretne terminy postępowania.
Poniżej „protokół” EU-RHAB wraz z opracowaniem oraz EpSSG NRSTS (2005 r.) wraz z opracowaniem:- EU-RHAB 2016 V5 – https://rhabdoid.info/wp-content/uploads/2021/12/EU-RHAB-Protokoll-Stand-08.12.2016.pdf
- Opracowanie EU-RHAB Clinical and genetic risk factors define two risk groups of extracranial malignant rhabdoid tumours (eMRT/RTK) – https://rhabdoid.info/wp-content/uploads/2021/12/rabdoides.pdf
- EpSSG NRSTS 2005 2014 V1.2 – https://rhabdoid.info/wp-content/uploads/2022/01/EpSSG-NRSTS-version-1-2.pdf
- Opracowanie EpSSG NRSTS Outcome of extracranial malignant rhabdoid tumours in children registered in the European Paediatric Soft Tissue Sarcoma Study Group Non-Rhabdomyosarcoma Soft Tissue Sarcoma 2005 Studyd EpSSG NRSTS 2005 – https://rhabdoid.info/wp-content/uploads/2021/12/4-_Brennan__2016_Extra_Cranial_NRSTS.pdf
- Opracowanie praktyczne EpSSG NRSTS na bazie doświadczeń jednego ośrodka – Rhabdoid tumor of the liver: Report of 6 pediatric cases treated at a single institute – https://www.semanticscholar.org/paper/Rhabdoid-tumor-of-the-liver%3A-Report-of-6-pediatric-Cornet-Lambert/db82fa596e4ce64afa8f187cc1f9c6eaaf05e92e
- Chemioterapia składa się cykli i bloków. EU-RHAB proponuje dwa lub trzy takie bloki w zależności od kluczowej decyzji czy będzie stosowana chemia wysokodawkowa HDCT z HSCT/ASCT/ASCR (o tym dalej). Każdy z zaproponowanych bloków dzieli się na trzy cykle:
- DOX – doksorubicyna (Doxorubicin) również pod nazwą adriamycyna,
- ICE – ifosfamid (Ifosfamide), karboplatyna (Carboplatin), etopsyd (Etopside) również pod nazwą VP-16,
- VCA – winkrystyna (Vincristine), cyklofosfamid (Cyclophosfamide), aktynomycyna D (Actinomycyn D).
- Jeżeli z jakichś ważnych powodów nie jest z góry planowana radioterapia (tylko po szczegółowej konsultacji w referencyjnym ośrodku radioterapii!!!!) bo wiek nie pozwala (chociaż wiek nie jest żadnym przeciwskazaniem w obliczu tej choroby stąd potrzeba każdorazowej oceny radioterapeuty!!!!) albo jest planowany przeszczep wtedy należy zmodyfikować cykle i najlepiej zdecydować się na VDC/ICE, VDCy/IE (https://www.ejcancer.com/article/S0959-8049(21)01183-7/ https://www.semanticscholar.org/paper/Rhabdoid-tumor-of-the-liver%3A-Report-of-6-pediatric-Cornet-Lambert/db82fa596e4ce64afa8f187cc1f9c6eaaf05e92e) lub VDC/CyCE z tej prostej przyczyny, że EU-RHAB zakłada obowiązkową radioterapię lokalną, regionalną oraz w ramach przerzutów przy jednoczesnym podawaniu chemioterapii (radio-chemioterapia) stąd wydziela DOX do oddzielnego cyklu aby było możliwe stosowanie radioterapii dwa tygodnie po jej podaniu i cztery tygodnie przed kolejnym cyklem DOX. Z uwagi na zwiększoną toksyczność w przypadku objęcia obszarem naświetlania serca w innym przypadku w razie potrzeby jest wymagana konsultacja radioterapeuty. W przypadku gdy takie wydzielenie nie jest potrzebne należy stosować inne reżimy nie przedłuzające przerw pomiędzy pozostałymi cyklami – bazując na doświadczeniu danej placówki i konsultacji z innymi placówkami.
- PRAKTYKA!!!! Istnieją doniesienia i praktyczne zastosowanie (np. przez lekarzy CZD) potwierdzone również w opracowaniach lezenia AT/RT, RTK, guza Wilmsa o skutecznym działaniu samego reżimu ICE gdzie ifosfamid wydaje się mieć kluczowe znaczenie. Są również proponowane jak wyżej (Boston Childrens Hospital/Dana-Farber Cancer Institute – BCH, Beijing Tongren Hospital of China Capital Medical University – BTH) intensywne (wyższe dawki) VDC/ICE a dodatkowo BTH proponuje zmianę na AVCP (doksorubicuna – andriamycyna – antracykliny, winkrystyna, cisplatyna, cyklofosfamid) / VIDE (winkrystyna, pirarubicyna – antracykliny, ifosfamid, etopsyd) przy braku spodziewanej odpowiedzi (https://rhabdoid.info/wp-content/uploads/2022/05/CMAR-309274-case-analysis-of-14-children-with-malignant-rhabdoid-tumor-o.pdf). Inne donoszą o skuteczności wysokich dawek akylatorów (cyklofosfamid) inne o doksorubicynie (antracykliny), jeszcze inne o dodaniu aktynomycyny D np:
- Surgery and Actinomycin Improve Survival in Malignant Rhabdoid Tumor – https://rhabdoid.info/wp-content/uploads/2021/12/SRCM2013-315170.pdf
- High Dose Alkylator Therapy for Extracranial Malignant Rhabdoid Tumors in Children – https://rhabdoid.info/wp-content/uploads/2021/12/Pediatric-Blood-Cancer-2014-Venkatramani-High-dose-alkylator-therapy-for-extracranial-malignant-rhabdoid-tumors-in.pdf
- informacje również w pozostałych wymienionych opracowaniach.
Ta różnorodność działających lub nie chemioterapeutyków wynika z faktu polimorficznej zróżnicowanej morfologi RT, wieku pacjentów i stosowanych dawek leków, lokalizacji ogniska pierwotnego, czasu i długości podawania leków (cykle komórkowe), stosowania radioterapii, która może być bardziej skuteczna w skojarzeniu z pewnymi lekami, zróżnicowania kombinacji lekowych, historycznych danych i kwalifikacji pacjentów, ilości samej próby badanej. W tak skomplikowanej wzajemnie odziaływującej mieszanienia elementów o niewielkiej próbie nie jest praktycznie możliwe wskazanie jednego działającego schemtau stąd tylko możliwość statystycznego wskazania na zasadzie prawdopodobieństwa sposobów postępowania. Dlatego stosowanie chemioterapii wymaga od lekarza praktycznej wiedzy, dociekliwości i świadomości tych ograniczeń. Praktyka pozwoli działać metodycznie i skutecznie zamiast przypadkowo lub wcale w momencie gdy będzie to najbardziej potrzebne.
- WAGA / DAWKOWANIE!!!! Dawkowanie generalnie jest przedstawione w odniesieniu do powierzchni ciała BSA. W EU-RHAB dla młodszych dzieci poniżej 6mc lub poniżej wagi 10kg dawki są przedstawione w odniesniu do proporcji wagowej BW. Jest to ważne z dwóch względów i o ile zmniejszenie dawek jest wprost ujęte w EU-RHAB w przypadku wystąpienia krytycznych efektów ubocznych (wraz z opisem jak je opanować) tak dla dzieci z pogranicza 9/10kg róznice w dawkowaniu dochodzą do 30%!!!! i mogą powodować nieskuteczność terapii. To jest kluczowa sprawa jaką należy przedyskutować z lekarzem prowadzącym w przypadku braku spodziewanych spadków parametrów krwi – zwłaszcza będzie to widoczne przy pierwszym bloku ICE przygotowującym do proceduty pobrania komórek macierzystych. Jeżeli wystąpi taka sytuacja w połączeniu z brakiem typowych objawów spodziewanych po chemioterapii jak nudności, brak apetytu, złe samopoczucie itp. a w szczególności brak spodziewanych spadków ilości białych krwinek krwi w tym głownie neutorfili prawdopodobnie mamy do czynienia z brakiem indywidualnego dopasowania dawek chemioterapeutyków. Należało by zatem zaniechać stosowania proporcji wagowej na rzecz tabelarycznie skorygowanych dawek opartych o alogrytm BSA – https://ascopubs.org/doi/abs/10.1200/JCO.2016.34.15_suppl.10546
- INTENSYFIKACJA!!!! Dla młodszych pacjentów wpada się w zamknięty krąg potencjalnego niepowodzenia gdyż redukuje się dawki z powodu potencjalnej toksyczności ze stratą dla potencjalnej przeżywalności – ten temat jest poruszany w opracowaniach i warto go omówić z lekarzem dostosowując dawki indywidualnie zaczynając od mniejszych i zwiększając w miarę leczenia (np. zaczynając od zredukowanej dawki wskazanej w „protokole” co 30% do osiągnięcia dawki docelowej bazującej na BSA). Opinia BCH podkreśla potrzebę intensyfikacji i eskalacj idawek chemioterapetyków tak bardzo jak to tylko jest tolerowalne – trzeba próbować i dostosowywać do aktualnego stanu pacjenta.
- Optymalna sytuacja to dobranie schematu chemioterapeutyków na podstawie wyników zleconych na początku badań ekspresji białek IHC i molekularnych szlaków – stosując tylko działające leki zamiast całego arsenału bazującego tylko na statystycznych analizach danych historycznych leczenia innych pacjentów. Pamiętajmy każdy przypadek jest indywidualny tak samo jak budowa i mikrośrodowisko guza.
- Zanim dojdziemy do konkretnych „protokołow” i ich nazw należy jasno powiedzieć, że jest to tylko schemat postępowania, zbiór dobrych praktyk, konsensus, opracowanie historycznych doświadczeń grup badawczych, złoty środek. W żadnym przypadku nie może być on traktowany dosłownie a zwłaszcza przez ośrodek nie mający do czynienia z takimi przypadkami na co dzień. Każdy przypadek należy traktować indywidualnie, dostosowująć w jak najlepszej wierze do określonego pacjenta, jego aktualnych i przyszłych potrzeb zapewniając wzrost szans na długie przeżycie. W dokumentach tych jest tak wiele niuansów, powtórzeń, znaków zapytania wynikających z prostego faktu – przypadków jest mało, rozpoznanie zwłaszcza historyczne mogło być wstępnie nieprawidłowe ze względu na trudność w rożnicowaniu, dość dynamiczny przebieg u młodszych pacjentów, możliwość występowania praktycznie w całym organiźmie, dobór środków i narzędzi leczniczych – zatem trudno wyróżnić jeden konkretny działający schemat leczenia w każdym przypadku a trudno zastosować taki ogólny plan bez jego precyzyjnego indywidualnego dostosowania.
- G-CSF
Jest cytokiną stymulująca wzrost kolonii granulocytów odpowiadająca za proliferaję, różnicowanie kriotwórczych prekursorów oraz aktywację neutrofili (obok GM-CSF stymulującą także makrofagi). Odpowiada również za migrację neutrofili do tkanek obwodowych. Stymuluje odpowiedź imunologiczną neutrofili oraz bezpośrednio i pośrednio odpowiedź pozostałych elementów układu immunologicznego. New insight into the mechanism of granulocyte colony-stimulating factor (G-CSF) that induces the mobilization of neutrophils
Jest to element intensywnych reżimów chemioterapii mający na celu przeciwdziałaniu jej skutkom tj. głownie niebezpiecznej neutropenii oraz jako element wysokodawkowej chemioterapii (HDCT) przy pozyskiwaniu komórek macierzystych (autologous stem cell rescue ASCR) i ich transplantacji (autologous stem cell transplantation ASCT).
Stosowanie jest elementem „protokołów” EU-RHAB, EpSSG NRSTS a także innych. Ma to na celu intensyfikację leczenia odnośnie dawek jak i skrócenia odstępu czasu w ich podawaniu zgodnie z teorią Log Cell Kill i dążenia do usunięcia jak największej ilości komórek nowotworowych z organizmu. Opis dawkowania jest szczegółowo zaprezentowany w protokołach i najczęściej rozpoczyna się dobę po i kończy dobę przed stosowaniem chemioterapeutyków – w czasie całego spodziewanego nadiru! W czasie pobrania komórek macierzystych stosowany jest kilka dni wcześniej (3-5) i stosowany przez cały czas ich pozyskiwania. W czasie wystąpienia neutropenii przez cały jej okres aż do otrzymania zadowalających wyników badań krwi.
Niestety G-CSF posiada cały szereg możliwych działań niepożądanych w tym bardzo poważnych jak białaczki i niebezpiecznych syndromów MDS/AML. Jednakże jego wpływ na skuteczność (intensyfikacja, łagodzenie skutków ubocznych) terapii wydaje się przewyższać potencjalne komplikacje.
Nie jest również dokładnie zbadane i poznane odziaływanie stosowania G-CSF na guzy lite. Niektóre badania dowodzą wręcz niekorzystny wpływ tej krwiotwórczej cytokiny (obok typowo zapalnych IL-1, IL-6, TNFα, TNFβ) na przebieg choroby a samą pierwotną ekspresję/produkcję G-CSF/G-CSFR przez komórki nowotworowe za skranie niekorzystną, zwieksząjąca proliferację, ekspansję i wpływające na mikrośrodowisko nowotworu. Dodatkowo podejrzewa się niekorzystny wpływ na tworzenie się nowych włosowatych naczyń krwionośnych – angiogenezę – redukującą na przykład dobroczynny wpływ radioterapii.
The role of granulocyte colony‑stimulating factor in breast cancer development: A review
G-CSF and G-CSFR are highly expressed in human gastric and colon cancers and promote carcinoma cell proliferation and migration
Badania na podgrupie wydzielonych linii komórek macierzystego typu neuroblastomy Cancer Stem Cell-like (CSC) wykazały, że ekspresja receptora G-CSF przez te linnie komórkowe oraz stosowanie w terapii G-CSF prowadzi do eskpansji, przyśpiesza wzrost nowotworu oraz przerzutów poprzez aktywację czynnika transkrypcyjnego STAT3. Czynnik ten podejrzewa się też o wpływ na przebieg choroby w RT.
G-CSF promotes neuroblastoma tumorigenicity and metastasis via STAT3-dependent cancer stem cell activation
Histone deacetylase inhibitor panobinostat induces antitumor activity in epithelioid sarcoma and rhabdoid tumor by growth factor receptor modulation
Biorąc pod uwagę te infomacje rozważa się stosowanie blokady pętli sygnałowej G-CSF/STAT3 poprzez używanie przeciwciał G-CSF lub inhibitorów STAT3 zmniejszając subpopulację komórek CSC, prowadząć do regresji, blokująć przerzuty i zwiększająć chemiowrażliwość. Badania te sugerują traktowanie G-CSF jako czynnika nowotworowego (przynajmniej w neuroblastomie) i sugeruje ponowną przekrojową ocenę jego stosowania. Dodatkowo jest bardzo ograniczona liczba dowodów na zwiększone przeżycie pacjentów przy stosowaniu G-CSF jako wspomagania chemioterapii.
Optymalnym podejściem było by sklasyfikowanie danego przypadku w zakresie aktualnie wyróżnionych grup RT – badając szczegółowo charakterystykę nowotworu. Jeżeli charakterystyka transkrypcyjna jest na bazie układu immunologicznego z odpowiedzią zapalną (większość pozanerkowych przypadków) to rozsądnym było by stosowanie G-CSF wybiórczo (tak jak innych cytokin pro-zapalnych), rezerwując jes tylko do ASCR/HDCT/ASCT. Dodatkowo stosując leczenie/suplementy przeciwzapalne zamiast wzbogacania terapii o elementy mogące sprzyjać ogólnemu dodatkowemu zapaleniu organizmu.
Leczenie wybiórcze mogło by polegać na wybraniu/modyfikacji reżimu chemioterapii rezerwującej „dodatkowy” czas w ramach cyklu ale bez zwiększania czasu samego bloku leczenia. Na przykładzie EU-RHAB i bloku składającego się z 3 cykli – DOX-ICE-VCA, każdy w 2-u tygodniowych interwałach dających łącznie 6 tygodni na blok przy zastosowaniu G-CSF. Modyfikacja takiego bloku polega na dołączeniu DOX do VCA z pominięciem lub nie aktynomycyny D stosując standardowy blok VCD-ICE w 3-y tygodniowych interwałach dający sumarycznie też 6 tygodni na blok bez stosowania G-CSF. Dodatkowo standardowe bloki VCD-ICE przwidują wyższe dawki każdego z chemioterapeutyków mogące dochodzić nawet do zwiększenia o 30-50% względem EU-RHAB co może przyczynić się do większej kontroli porcesu nowotworowego. - Radioterapia
- Jest kluczowym jeżeli nie najważniejszym elementem terapii zaraz po interwencji chirurgicznej. Cały „protokół” EU-RHAB jest zbudowany wokół niej zakładając równoczesność stosowania z chemioterapią w odpowiedni sposób modyfikując poszczególne składniki cykli podawania chemioterapeutyków. Również z większości opracowań (z poprzednigo punktu EU-RHAB, EpSSG NRSTS) wskazuje się statystyczną pewność radioterapii jako predyktora dłuższego przeżycia i mniejszej śmiertelności. Trudność w ocenie wynika z kilku faktów:
- analizy operają się na danych historycznych retrospektywnych (starsze rodzaje sprzętu do radioterapii, inne dawki),
- część z nich porównuje różne schematy leczenia,
- dodatkowo w opracowaniach zebrane są wszystkie lokalizacje wystepowania nowotworów z ewentualnym wyłączniem AT/RT,
- z powodu małej liczby przypadków,
- tendencji do stosowania u starszych pacjentów,
- i dla pacjentów o znacznym stopniu zawansowania.
- WIEK!!!! Tak jak w stosowaniu chemioterapii dla najmłodszych pacjentów pojawia się magiczna liczba 10kg tak dla radioterapii pojawia się szereg takich liczb dla wieku: 3 lata, 2 lata, 18 miesięcy, 12 miesięcy, 6 miesięcy. Lekarze i „protokoły” posługują się tymi liczbami na zasadzie paradygmatów i oceny niewielkiej ilości danych historycznych – to ważne dla historycznych zastosowań starszych modeli urządzeń do napromieniania. Dzisiejsze urządzenia (IMRT – modulacja dawki, VMAT – modulacja z obrotem głowicy, protonoterapia) pozwalają na duże skrócenie całego cyklu napropieniania, większą kontrolę obszaru naświeltnia, rozkładu podawanych dawek promieniowania, jednoczasowe naświetlanie większego obszaru z podaniem dodatkowych dawek w wybranych podobszarach (boost), kształtu tych dawek. Dlatego należy ją stosować jak najwcześniej, nie bać się czekając na przekroczenie magicznego wieku bo jeszcze się nic nie dzieje albo zmiana nie wymaga jeszcze naświetlania. Należy każdorazowo przeanalizować ryzyko i korzyści ponieważ to czy radioterapię można było zastosować wcześniej dowiemy się tylko po fakcie…
- EU-RHAB podaje 18 miesięcy jako magiczną liczbę, referencyjne ośrodki polskie naświetlają już 12 miesięczne dzieci, zagranioczne nawet 6 miesięczne.
- Przy zastosowaniu EU-RHAB zastosowanie radioterapii jest:
- obowiązkowe i to w jak najszbyszym możliwym terminie,
- przy jednoczesnym podowaniu chemioterapii,
- naświetla się lożę po guzie,
- sam guz gdy nie jest możliwy doszczędny zabieg chirurgiczny,
- lokalny obszar gdy choroba jest zlokalizowana (węzły chłonne),
- obszar lub region po chirugicznie usuniętych przerzutach,
- obszar lub region po całkowitej odpowiedzi na chemioterapię przerzutów.
- Dla młodszych pacjentów wpada się poraz kolejny w zamknięty krąg potencjalnego niepowodzenia gdyż odsuwa się w czasie radioterapię z powodu potencjalnej toksyczności ze stratą dla potencjalnej przeżywalności – ten temat jest poruszany w opracowaniach i warto go omówić z lekarzem dostosowując najbardziej korzystny sposób leczenia zakładając dodatkowo radioterapię jako kluczowy czynnik statystyczny.
- Oczywiście efekty uboczne jak przy każdej terapii mogą wystąpić jednak należy odróżnić naświeltanie nowoczesną techniką ograniczonego obszaru niewielkiej częsci organu po interwencji chirurgicznej wraz z ewentualnymi węzłami chłonnymi od:
- naświetlania całego mózgu,
- całych płuc,
- całej jamy brzusznej,
- dużych struktur kostnych
o czym najczęsciej traktują opracowania w kontekście efektów ubocznych i czego najczęściej obawiają się lekarze onkolodzy dlatego wymagana jest wczesna konsultacja radioterapeuty.
- Late Effects of Abdominal Irradiation in Children: A Review of the Literature – https://rhabdoid.info/wp-content/uploads/2022/01/227.full_.pdf
- Ocena skojarzonego leczenia przeciwnowotworowego ze szczególnym uwzględnieniem radioterapii u dzieci z rozpoznaniem guzów tkanek miękkich – https://rhabdoid.info/wp-content/uploads/2022/01/index.pdf
- Jest kluczowym jeżeli nie najważniejszym elementem terapii zaraz po interwencji chirurgicznej. Cały „protokół” EU-RHAB jest zbudowany wokół niej zakładając równoczesność stosowania z chemioterapią w odpowiedni sposób modyfikując poszczególne składniki cykli podawania chemioterapeutyków. Również z większości opracowań (z poprzednigo punktu EU-RHAB, EpSSG NRSTS) wskazuje się statystyczną pewność radioterapii jako predyktora dłuższego przeżycia i mniejszej śmiertelności. Trudność w ocenie wynika z kilku faktów:
- Wysokodawkowa chemioterapia
From November 2010, all extra-cranial MRT patients who achieved complete remission after conventional therapy underwent HDCT/ASCR with uniform conditioning with melphalan, etoposide, and carboplantin. - Chemioterapia standardowa ratunkowa
Na bazie doświadczeń EpSSG NRSTS 2005 2014 V1.2 – https://rhabdoid.info/wp-content/uploads/2022/01/EpSSG-NRSTS-version-1-2.pdf w zakresie leczenie mięsaków można przedstawić zaprezentowane tam terapie ratunkowe – drugiej linii:- Vinorelbine,
- Wysokie dawki ifosfamidu > 12-6 g/m2 (z doxorubicyną),
- Gemcitabina,
- Gemcitabina i docetaxel,
- Gemtabicyna i Vinorelbina,
- Taksany – paclitaxel,
- Trabectidina,
- Imatinib,
- Sorafenib,
- Inhibitory rapamycyny (mTor),
- Temodal-toptecan.
- Multimodalna terapia
Przeglądając poszczególne przypadki analizujące użycie HDCT/HSCT oraz radioterapii wskazują jednocznacznie, że zdecydowana większość przeżyć odległych jest osiągana dla zastosowania przynajmniej jednego lub obydwu z powyższych:- Extracranial Malignant Rhabdoid Tumors in Childhood The Childrens Hospital Los Angeles Experience – https://rhabdoid.info/wp-content/uploads/2021/12/Cancer-2007-Madigan-Extracranial-malignant-rhabdoid-tumors-in-childhood.pdf
- Extra-cranial Malignant Rhabdoid Tumor in Children: A Single Institute Experience – https://rhabdoid.info/wp-content/uploads/2021/12/crt-2013-176.pdf
- High Dose Alkylator Therapy for Extracranial Malignant Rhabdoid Tumors in Children – https://rhabdoid.info/wp-content/uploads/2021/12/Pediatric-Blood-Cancer-2014-Venkatramani-High-dose-alkylator-therapy-for-extracranial-malignant-rhabdoid-tumors-in.pdf
- Ewaluacja
Wedłu dostępnych na dzień dzisiejszy ogólnych opracowań dużych grup pacjentów z podstawowym podziałem na AT/RT, RTK/eMRT bez rozróżnienia na konkretne lokalizacje anatomiczne (w tym wątroba) wyróżnia się starszy wiek pacjenta (powyżej 12-u lub 24-ech miesiący), niższe stadium zaawansowania choroby, brak mutacji dziedzicznych (SMARCB1/SMARCA4), resekcja kompletna, zastosowanie radioterapii jako główne predyktory przeżycia.
Same statsytyki przeżycia wyglądają następująco (Current and Emerging Therapeutic Approaches for Extracranial Malignant Rhabdoid Tumors):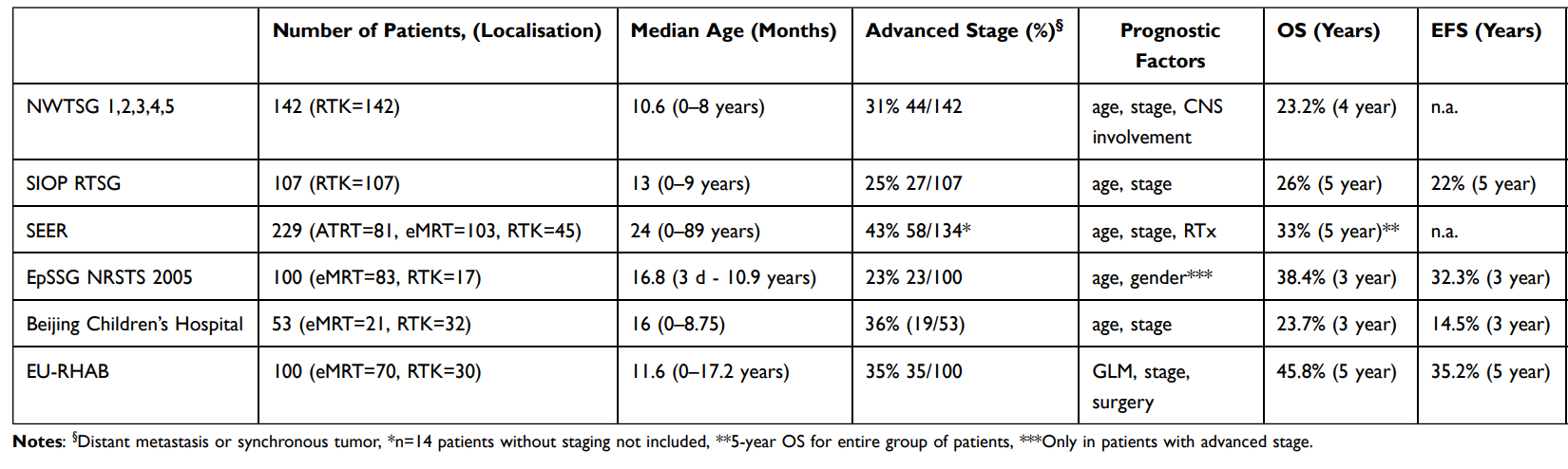
Jak widać wyniki całkowitego przeżycia mają charakter mieszany oscylujący w zakresie 23,2% do 45,8%. Ma to związek z tym, że bazują na różnych proporcjach grup pacjentów, w różnym stanie i leczonych różnymi „protokołami”. Najbardziej zbliżone i poprawiające się są wyniki EpSSG NRTS oraz EU-RHAB gdyż są to w miarę podobne podejścia prowadzenia pacjentów w zuniformizowany, intensywny sposób z dużym naciskiem na jak najszybsze stosowanie radioterapii. - Scenariusze
- Ośrodki zagraniczne/konsultacje:
- Europa
- EU-RHAB – Prof. Dr. Dr. Michael C. Frühwald; Klinik für Kinder und Jugendliche, Klinikum Augsburg – michael.fruehwald@klinikum-augsburg.de
- EpSSG – Dr. Bernadette Brennan; Royal Manchester Children’s Hospital – bernadette.brennan@cmft.nhs.uk
- University Hospital Tübingen – https://www.medizin.uni-tuebingen.de/ipu/?lang=en
- Institut Curie Paris – https://international.patient.curie.fr/?lang=en
- Hôpital Bicêtre AP-HP Paris – https://hopital-bicetre.aphp.fr/contact/
- USA:
- St. Jude Children’s Research Hospital Memphis – https://www.stjude.org/patient-referrals/seek-treatment.html
- Boston Childrens Hospital/Dana-Farber Cancer Institute – https://www.danafarberbostonchildrens.org/stay-connected/second-opinions
- Memorial Sloane Kettering Cancer Center New York – https://www.mskcc.org/appointments/refer-patient
- The Childrens Hospital Los Angeles – https://www.chla.org/refer-patient
- Inne:
- Beijing Tongren Hospital of China Capital Medical University – huamgdomgshemg@163.com
- Ankara Atatürk Training and Research Hospital/Children’s Hematology and Oncology Hospital – saytac1@gmail.com
- Seoul National University Children’s Hospital and Cancer Research Institute – hyshin@snu.ac.kr
- Europa
- Leczenie „niestandardowe” – testy kliniczne (clinical trials)
Z uwagi na to, że aktualnie genetyczna naprawa genów promotorowych jest niemożliwa stosowana jest alternatywna strategia – indukowania różnicowania morfologicznego metodami farmakologicznymi stąd wiele zaporponowanych terapii pomujących podatność nowotworów embrionalnych na celowane modyfikatory epigenetyczne starając się wytworzyć trwałą nieodwracalną blokadę wzrostu:- Checkpoint Inhibitors: This class of drugs is of increasing interest in the treatment of MRT. Rhabdoid tumors have been shown to have a very low tumor mutational burden and no evidence of MSI high status, however, despite this, there is emerging evidence to support that specific subsets of rhabdoid tumors, particularly the extra-cranial ones, are highly infiltrated with cytotoxic T cells and overexpress immune checkpoint markers and therefore are likely to be sensitive to immunotherapeutic targeting with immune checkpoint inhibitors. DFCI/BCH has an open trial for any INI1 deficient solid tumor (NCT04416568). This would be a reasonable consideration. These are also agents that are likely to be available outside a clinical trial, and that have fairly well understood administration guidelines and toxicity profiles. If clinical trial enrollment not possible, one couldconsider giving these off-label at your institution.
- GPC3 Immunotherapy: Approximately half of all malignant rhabdoid tumors express Glypican 3 (GPC3) Dr. Michael Ortiz, at Memorial Sloane Kettering (MSK) is an expert in pediatric renal tumors, malignant rhabdoid tumors, and novel therapeutics. MSK has a current phase 1 study of a GPC3 directed monoclonal antibody, Codrituzumab, at MSK (NCT04928677), lower age limit is 12 months. There is a plan to open this at DFCI/BCH within the year. This drug would not be available outside of a clinical trial.
- XPO1 Inhibitors: Rhabdoid tumors have been shown in preclinical trials to be dependent upon the nuclear pore exportin 1 (XPO1) and thereby are hypersensitive to its inhibition. There was a COG study, ADVL 1414, which was a phase 1 study of XPO1 inhibitor Selinexor, is now closed except to high grade gliomas. The drug may be accessible via other means such as compassionate access and conventional means since FDA approved for other indications. Karyopharm is the company that manufactures this drug and has a strong interest in providing new therapies for rare pediatric tumors, and has at times made the drug available in tablet and liquid form.
- Aurora Kinase Inhibitors: Alisertib is an AURKA inhibitor which has shown promise for the treatment of ATRT, a closely related, but not identical tumor, both in a case series and preliminarily as a phase II study. There is a multicenter study (NCT02114229) open at several locations. I believe that the closest site would be Children’s National. This drug is likely only available on a clinical trial. It is important to recognize that the promising results in ATRT will not necessarily translate into good effect in MRT.
- MDM2 Inhibitors: Idasanutlin is an MDM2 inhibitor which showed promise in a preclinical rhabdoid tumor study and would be potentially available in clinical trial setting as a monotherapy or in combination with cyclo/topo (NCT04029688). This builds on the theoretical approach of lowering apoptotic potential of malignant cells by adding a sensitizing agent. Dr Dave Shulman is in our Developmental Therapeutics division and has a MDM2 inhibitor trial at DFCI/BCH
- CDK4/6 Inhibitors: SMARCB1 normally represses cyclin D-CDK4/6 through direct transcriptional repression of CCND1 and upregulation of genes encoding the cyclin D-CDK4/6 negative regulators, p21Cip1 and p16INK4A. Biallelic loss of SMARCB1 results in altered expression of key cell-cycle regulators and reversal of cell-cycle arrest. There is a trial of Palbociclib + chemo combination trial (NCT03709680) which has been open at DFCI/BCH.
- Proteasome Inhibitors: Several research studies have suggested that protein synthesis in rhabdoid tumors can be therapeutically leveraged by inhibiting the proteasome. Proteosome inhibitors have been combined with chemotherapy agents (such as the MSK POETIC study combining the proteasome inhibitor Carfilzomib with Cyclophosphamide and Etoposide (NCT02512926), or Bortezomib. an earlier generation proteasome inhibitor has been given with other chemotherapeutics.
- Decitabine: Clinical evidence for a biological effect of epigenetically active decitabine in relapsed or progressive rhabdoid tumors (https://rhabdoid.info/wp-content/uploads/2022/05/Pediatric-Blood-Cancer-2021-Steinb-gl-Clinical-evidence-for-a-biological-effect-of-epigenetically-active-decitabine.pdf).
- Current and Emerging Therapeutic Approaches for Extracranial Malignant Rhabdoid Tumors (https://rhabdoid.info/wp-content/uploads/2022/05/CMAR-289544-current-and-emerging-therapeutic-approaches-for-extracranial.pdf) sumaryczne podsumowanie powyższych uzupełnione o aktualne badania:
- Epigenetic Inhibitors (HDACi, DNMT, EZH2…),
- Cell Cycle Inhibitors (CDK4/6 Cyclin D1 inhibitor),
- Kinase Inhibitors (Aurora A kinase inhibitor, mTORC1/2, PDGFR/FGFR, EGFR/HER2, VEGF, …),
- Pathway Specific Compounds (BMP, Wnt/beta-catenin inhibitor, Oncolytic virus, …),
- Immunotherapy (CAR-T-cell, PD-L1/PD-1 inhibitor),
- Other (ALDH, LOX, MDM2, MDM4, MDMX, Proteasome inhibitors, …).
- Kontakt
info@rhabdoid.info